Endpoints are needed throughout the vaccine development life cycle. Their selection are critically important and impact study outcomes. Each development stage needs a careful assessment of fit-for-purpose endpoints. In this article we will discuss how to develop, assess and select endpoints at each development step.
Development of pharmaceutical products requires generation of data that enable a comprehensive evaluation of benefit against potential risks. Prophylactic vaccines are no exception. The benefit component must be accurately measured throughout the product development life cycle to determine vaccine efficacy (VE). Measuring VE is accomplished with disease- and/or pathogen-specific clinical study endpoints. These endpoints are carefully tracked, captured, evaluated in detail and sometimes adjudicated to assess if study efficacy objectives are being met. This comprehensive approach is critical for regulators, public health experts, prescribers and patients to make informed decisions on licensing, guideline recommendations, clinical use, and patient adoption and adherence.
A vaccine benefit-risk profile relies on conclusions drawn from the analysis of VE endpoints.
Measuring efficacy endpoints is required for all novel vaccines and for existing vaccines used in new target populations, unless there are established and validated surrogates of efficacy.
From polio and influenza — against which the first vaccines demonstrated their efficacy decades ago — to more recent disease targets such as SARS-CoV-2 and RSV, the use of carefully defined endpoints is a fundamental part of assessing VE in both large, prospective, randomised, placebo-controlled and real-world studies.
The type of endpoints of interest varies throughout the clinical development life cycle.
Epidemiologists and clinicians are instrumental in assessing the key medical needs that a novel vaccine intends to address, as well as the targeted outcomes it hopes to achieve. Endpoints typically are considered across four dimensions:
1. Clinical triggers—The symptoms of the disease or ailment that will trigger further diagnostic procedures (clinical trial setting) or may form the basis for a diagnosis based on clinical judgment or other circumstantial evidence (real-world setting).
2. Pathogen identification—Currently dominated by high-throughput polymerase chain reaction (PCR) based on assays but, depending on the pathogen, other lab tests may play an important role, such as virological, microbiological cultures.
3. Geography and seasonality—Factors that influence when and where a disease outbreak is likely and how this impacts the positive and negative predictive values of any assays.
4. Population specifics—How different populations (e.g., infants vs. the elderly) express signs or symptoms differently, and how acceptability and application of diagnostic procedures may vary between these populations.
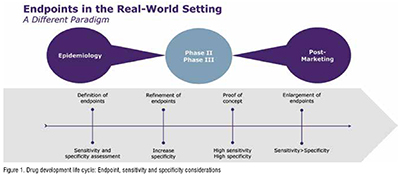
Throughout a clinical development plan, endpoints must be refined to strike the right balance between diagnostic sensitivity and specificity (Figure 1). When a test’s sensitivity is high, it is more likely to give a true positive result and correctly detect the presence of disease or illness. When specificity is high, it is more likely to give a true negative result and correctly identify the lack of disease or illness. Each has its place in the vaccine development life cycle, so both sensitivity and specificity should be assessed and their roles defined at the outset. Phase III studies require the highest sensitivity to ensure the entire disease burden is assessed for efficacy but also specificity as the vaccines are pathogen specific and sometimes restricted to strains or serotypes. (Figure 1)
The pre-clinical phase requires that experts assess the epidemiology of the disease in terms of incidence, prevalence, severity, risk factors, outcomes and other elements. Where feasible, disease burden and medical need are measured or assessed across different health care systems and geographies. These are typically assessed in non-interventional epidaemiologic studies, often called natural history or descriptive studies. The main objectives of a natural history study are to describe the following:
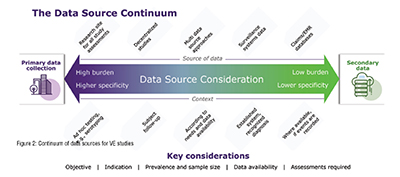
Natural history studies necessitate data that reflect the real-world management of patients and that their methodologies are different from clinical trials. There are many different data sources that can be used for such studies. Data can be generated for the purpose of the research in a specific setting or extracted from existing administrative data collection systems. Between these two extremes there is a continuum of different data sources, whose choice (based on granularity, quality, reliability) is also key to determine endpoints (Figure 2). Primary data collection is at one end of the spectrum and is generally time and cost intensive, representing a high burden with high specificity. Secondary data are a lower burden with respect to cost and time. However, the specificity is lower compared with primary data. (Figure 2)
When a new pathogen emerges, primary data need to be collected via a hospital- or site-based approach obtaining data from patients and their doctors, usually via questionnaires.
Primary data collection also can be useful for studying known pathogens, such as different pneumococcal serotypes in pneumococcal pneumonia CAP. In this case, as serotyping is not part of any existing surveillance system and is not routinely assessed in clinical practice, the information cannot be found in secondary data sources making primary data collection the best way to identify and track these specific patients.
At the other end of the spectrum, secondary data provide the opportunity to use existing data, such as insurance claims or infectious diseases surveillance systems.
Given all the factors that continually affect the generation of real-world data, it is important to create a nimble fit-for-purpose study design and to anticipate how the pathogen, the subjects, and any testing and treatment may change over time.
Endpoints captured in clinical development are precise endpoints with high specificity and high sensitivity across Phases II and III, such that precision peaks with highly targeted and reliable endpoints in Phase III.
Consider how one might track endpoints for an influenza vaccine. In Phase III, a diagnosis would be made according to a strictly defined set of criteria (e.g., cough, fever, shivering, general malaise, myalgia), but, to increase sensitivity of results, a PCR test would then be deployed to confirm viral presence. In this way, sensitivity (anyone who meets the symptom requirements) is combined with specificity (lab test) for optimal results.
Regulators also play an important role in ensuring optimal endpoints are used to properly reflect VE. Regulatory agencies publish general guidelines such as “Considerations for Developmental Toxicity Studies for Preventative and Therapeutic Vaccines for Infectious Disease Indications ” (2006). Guidance also may be much more targeted with pathogen-specific guidance, such as those developed for COVID-19, “Development and Licensure of Vaccines to Prevent COVID-19 ” (2020). While those examples come from the U.S. Food & Drug Administration other regions issue guidance similarly.
As a novel prophylactic vaccine moves into a post-marketing phase, endpoints of interest may shift to accommodate changing circumstances. With the experimental phases of development essentially over and with subjects no longer carefully selected according to stringent protocol defined criteria, the real-world setting brings new levels of complexity. For example, comorbidities tend to take on a much more significant role and endpoint sensitivity (increasing probability of detection) may take preference over specificity.
As more and more real-world evidence is generated, research experts will further use and adapt the endpoints to measure real-world vaccine effectiveness or post-marketing safety. It is also worth noting that less stringent endpoints—though still clinically relevant— also may be required to deliver meaningful insights and to inform vaccine policies. It is crucial that these endpoints also reflect standard of care and therefore the real-world methodology used to detect diseases/pathogens. For example, a recent study of the effectiveness of a bivalent COVID-19 vaccine booster was conducted in the Netherlands using self-reported COVID-19 as an endpoint. This example also reflects the fact that the timing of the study plays an important role in endpoint definition as pathogen-detection testing evolves over time.
Defining and measuring endpoints for novel prophylactic vaccine trials and real-world studies is a complicated but critically important piece of the development process. Case definitions that are overly restrictive may significantly impact incidence rates and detrimentally affect efficacy and success criteria. On the other hand, if case definitions are overly accommodating, they may introduce excess noise into the efficacy signal. From the outset, it is important to engage with the right experts and develop a thorough understanding of the targeted disease in order to accurately trace efficacy of the test solution.
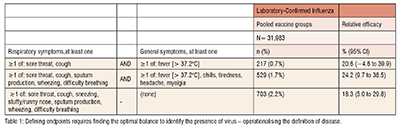
For example, Table 1 (adapted from “Efficacy of High-Dose Versus Standard-Dose Influenza Vaccine in Older Adults” ) shows results from a Phase III trial for an influenza vaccine. Here, a different combination of signs and symptoms leads to a dramatically different number of efficacy endpoints, all confirmed by lab testing. The figures shown here are all from the same study and show how by changing the qualifying symptoms has led to a threefold increase in case counts, and also impacted the lower bound of the confidence interval of VE. (Table 1)
Endpoint election is critically important and has a major impact on study outcomes and the assessment of VE. Selecting, defining and evaluating the endpoints to be used throughout the life cycle of vaccine development require a broad range of expertise from clinicians and epidemiologists. Optimal study development requires prospective planning to precisely define the disease and factors that may impact VE, from geography and seasonality to age, population and socioeconomic factors, as well as diagnostic methods. The success of a vaccine program will be determined by the endpoint measures and vaccine efficacy. A continuous dialogue with regulators on the endpoint selection is highly beneficial and increases the likelihood of positive regulatory reviews.

Paul Gillard serves as vice president and a medical lead for the vaccines therapeutic area within the PPD clinical research business of Thermo Fisher Scientific. In this role, he provides medical and scientific expertise with a focus on vaccine development strategies across various therapeutic areas. He joined the business in 2022 with training in internal medicine and more than 18 years of scientific leadership expertise in the clinical development of prophylactic vaccines.

Delphine Saragoussi serves as an executive director of the epidemiology and scientific affairs team of the real-world evidence (RWE) group within the PPD clinical research business of Thermo Fisher Scientific. As a physician, she specialises in public health and social medicine and has more than 20 years of applied experience in epidemiology and pharmacoepidemiology. Before joining the business, she served as a global therapy area lead in RWE and epidemiology at Lundbeck.

Sarah Rosen serves as a director within non-interventional studies, which is part of the RWE/peri- and post-approval practice within the PPD clinical research business of Thermo Fisher Scientific. Prior to joining the business, she was a project manager within lifecycle management at a global clinical research organisation and served as the observational subject matter expert. She has more than 15 years’ experience in project management and has successfully conducted a number of full-service noninterventional studies.













